目次
0. 再生処理におけるバリデーションとは
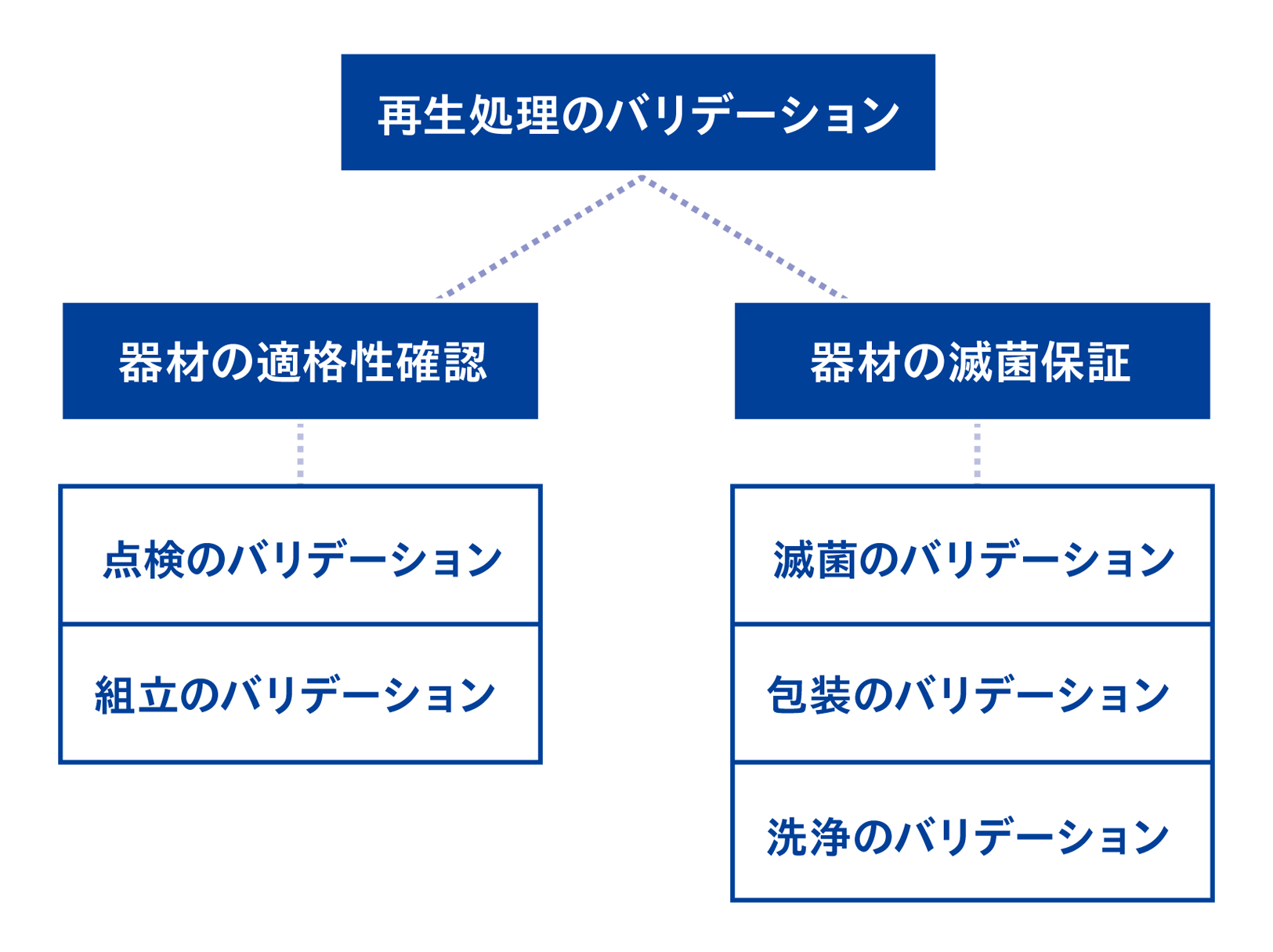
0-1. バリデーションとは ①器材の適格性確認と ②器材の滅菌保証を行うこと
再生処理におけるバリデーションとは、器材が患者さんに安全に使用できる状態になっているかを確認することです。
安全に使用できる状態とは、器材に破損がなく正常に機能する状態で、かつ滅菌条件の達成が保証された状態のことを言います。
破損がなく正常に機能する状態であるか確認することを「器材の適格性確認」、滅菌条件の達成を保証することを「器材の滅菌保証」と言い、この2つが再生処理のバリデーションを構成する2つの要件とされています。器材の適格性確認は、再生処理の工程のうち「点検」「組立」の工程で、器材の滅菌保証は「洗浄」「包装」「滅菌」の工程でバリデーションを行って確認します。
再生処理の各工程は、互いに繋がり、どれも欠くことのできない不可欠な工程です。再生処理のバリデーションも同様に、各工程のすべてのバリデーションを行ってはじめて実施できたと言うことができます。
0-2. ①器材の適格性確認:医療機器として本来もつべき品質・機能・安全性を確認する
器材の適格性確認は、「点検」「組立」の工程で行います。点検・組立の工程で必要なことは、器材が医療機器として本来もつべき品質・機能・安全性を備えているかを確認することです。錆や欠け、曲がりなどが発生してしまった器材を払い出すことのないよう、器材の適格性をきちんと確認する必要があります。

0-3. ②器材の滅菌保証:全ての器材において滅菌条件の達成を確認する
器材の滅菌保証とは、全ての器材において滅菌条件の達成を保証することです。「滅菌保証」と言うと、つい滅菌工程のみに目を向けてしまいがちですが、滅菌工程のみを検証しても滅菌保証は実現できません。器材が正しく洗浄されていなければ、滅菌剤が十分に曝露しません。また、正しく包装されていなければ、使用直前まで器材の無菌性を維持することはできません。滅菌保証をするためには、再生処理の工程の全てについて検証を行い、バリデーションをとる必要があります。
また、滅菌条件の達成は、器材表面だけでなく、内部まで確認する必要があります。一般的に、器材表面よりも器材内部のほうが滅菌しづらいと言われています。
例えば、下記のラパロ鉗子のような内腔器材の場合、外側よりも内腔のほうが滅菌しづらいです。したがって、インジケータ類を使用して滅菌確認をする場合、本来的にはラパロ鉗子の内腔にインジケータを設置する必要があります。

1. 高圧蒸気滅菌のバリデーションの流れ
本記事では、再生処理のバリデーションを構成する要件のひとつである「器材の滅菌保証」において、高圧蒸気滅菌の場合のバリデーションの流れを解説します。
1-1. 高圧蒸気滅菌のバリデーションは、IQ・OQ・PQの順に実施する
高圧蒸気滅菌器のバリデーションは3ステップで、IQ(据付時適格性評価)、OQ(運転時適格性評価)、PQ(性能適格性評価)の順番で実施します。これらの評価を実施する際は、高圧蒸気滅菌器の温度計や圧力計が適切にキャリブレーション(校正)されていることが前提となります。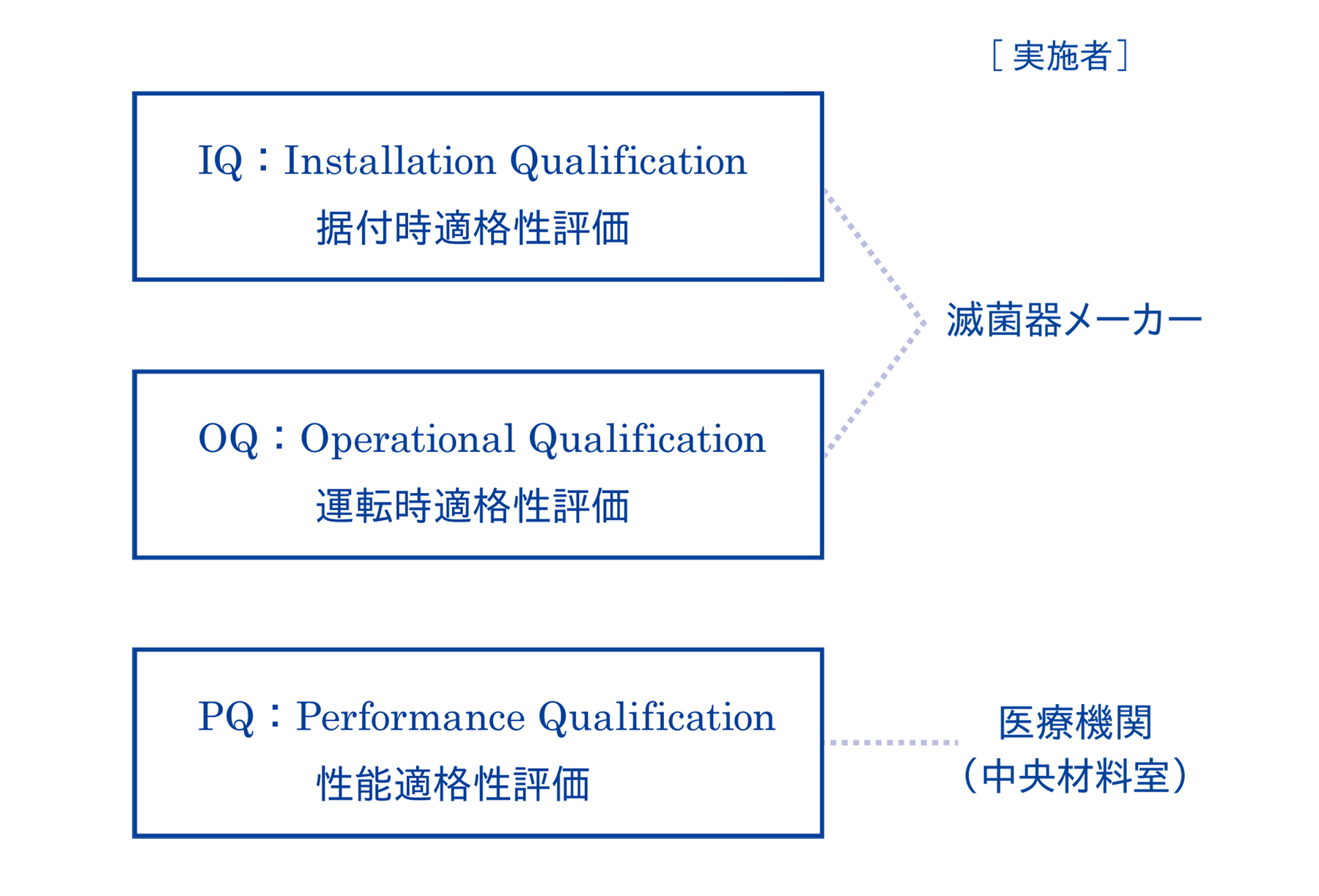
また、上記の工程が終了したら「標準作業手順書(SOP)」を作成します。日々の滅菌業務では常にSOPを基に作業を進めることにより、検証された工程の範囲内での滅菌作業を進めることができるようになります。
1-2. IQでは滅菌器の設置が正しく行われたかを確認する
高圧蒸気滅菌のIQでは、滅菌器の設置時に供給電圧や電力、蒸気圧力、吸水圧力、蒸気・吸水・排蒸気配管口径などの項目を検査します。
付属装置を含めて滅菌器が正しく設置されているかを確認します。
1-3. OQでは装置が正常に運転できる状態かを確認する
高圧蒸気滅菌のOQでは、温度や圧力センサーなどの正確性の確認や、滅菌器の圧力上昇速度、真空到達速度、真空到達度、温度や圧力の安定性、排蒸気速度、主要機器の動作確認などの項目を検査します。あわせて、コールドスポット(庫内で最も温度が上がりにくい場所)も特定します。
1-4. PQでは滅菌した器材が滅菌されているかを確認する
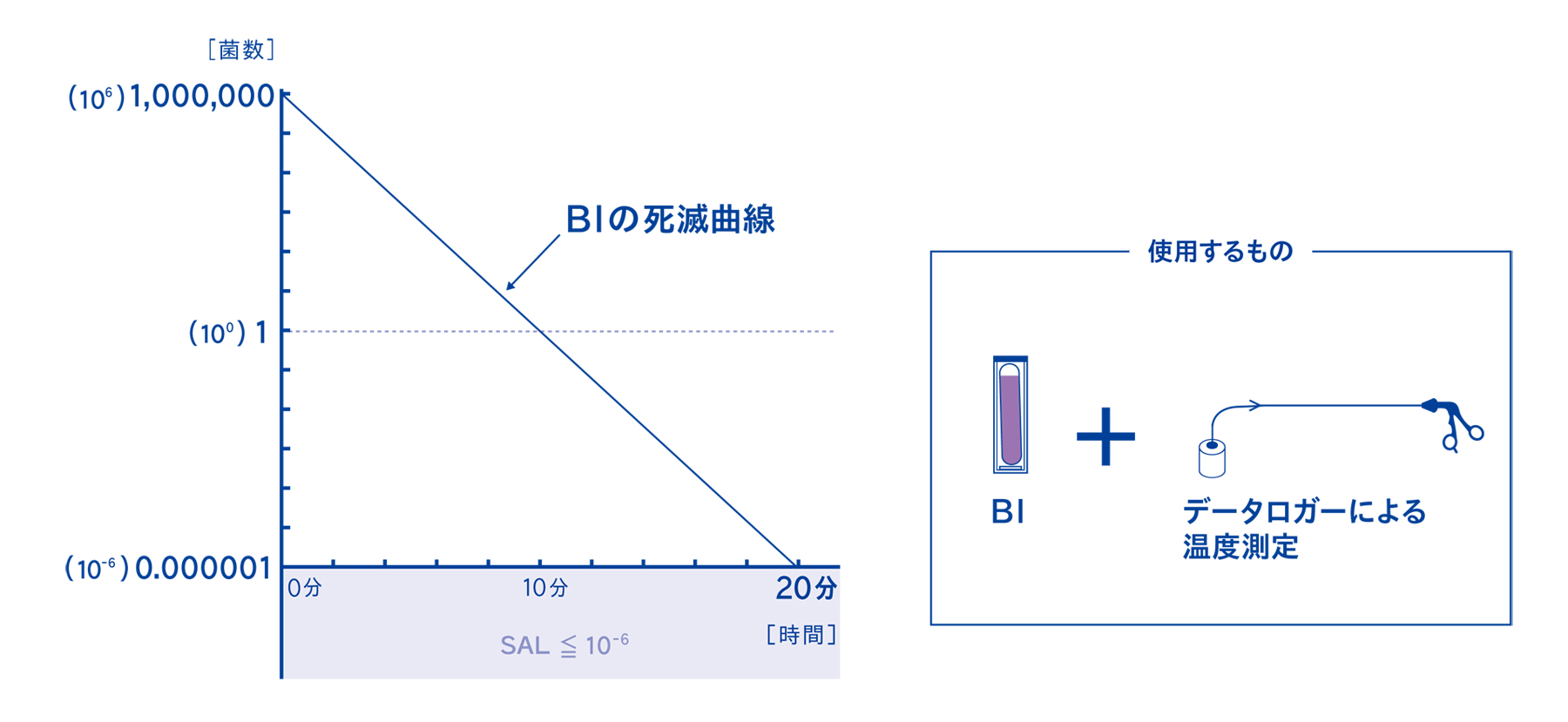
PQでは、最も滅菌が難しい器材である「マスター製品」を選定し、その滅菌確認を実施します。この時、器材の温度測定を行うデータロガーや、BIを用いて滅菌確認をします。
一般的に滅菌の条件が悪いとされる、積載量が最大の場合と、最小の場合の2つの条件においてマスター製品の滅菌確認を実施します。この時、滅菌条件の達成とあわせて、ドレインが発生していないか等、乾燥状態についても確認します。
 PQ実施中の様子
PQ実施中の様子
1-5. IQとOQは滅菌器メーカーが実施する
IQ・OQは、滅菌器メーカーでないと検証できない範囲のため、滅菌器メーカーが実施します。IQは据え付け時にのみ行いますが、定期点検ではOQの一部の検証の他、バリデーションの要である温度計や圧力計のキャリブレーションも実施してもらえるので、滅菌器メーカーに相談してみるのもおすすめです。
1-6. PQは医療機関(中央材料室)が実施する
医療機関ごとに、使用する器材やその包装形態、滅菌器への積載形態は異なります。そのため、PQは医療機関(中央材料室)が実施する必要があります。
PQを実施する主体は医療機関(中央材料室)ですが、滅菌器の設定も関わってくるため滅菌器メーカーが協力してくれることもあります。
2. 高圧蒸気滅菌のPQの流れ
2-1. 製品ファミリーの分類、マスター製品の選定、マスター製品の滅菌確認、の順に行う
高圧蒸気滅菌のPQは、まず「製品ファミリーの分類」、次に「マスター製品の選定」、最後に「マスター製品の滅菌確認」の順に行います。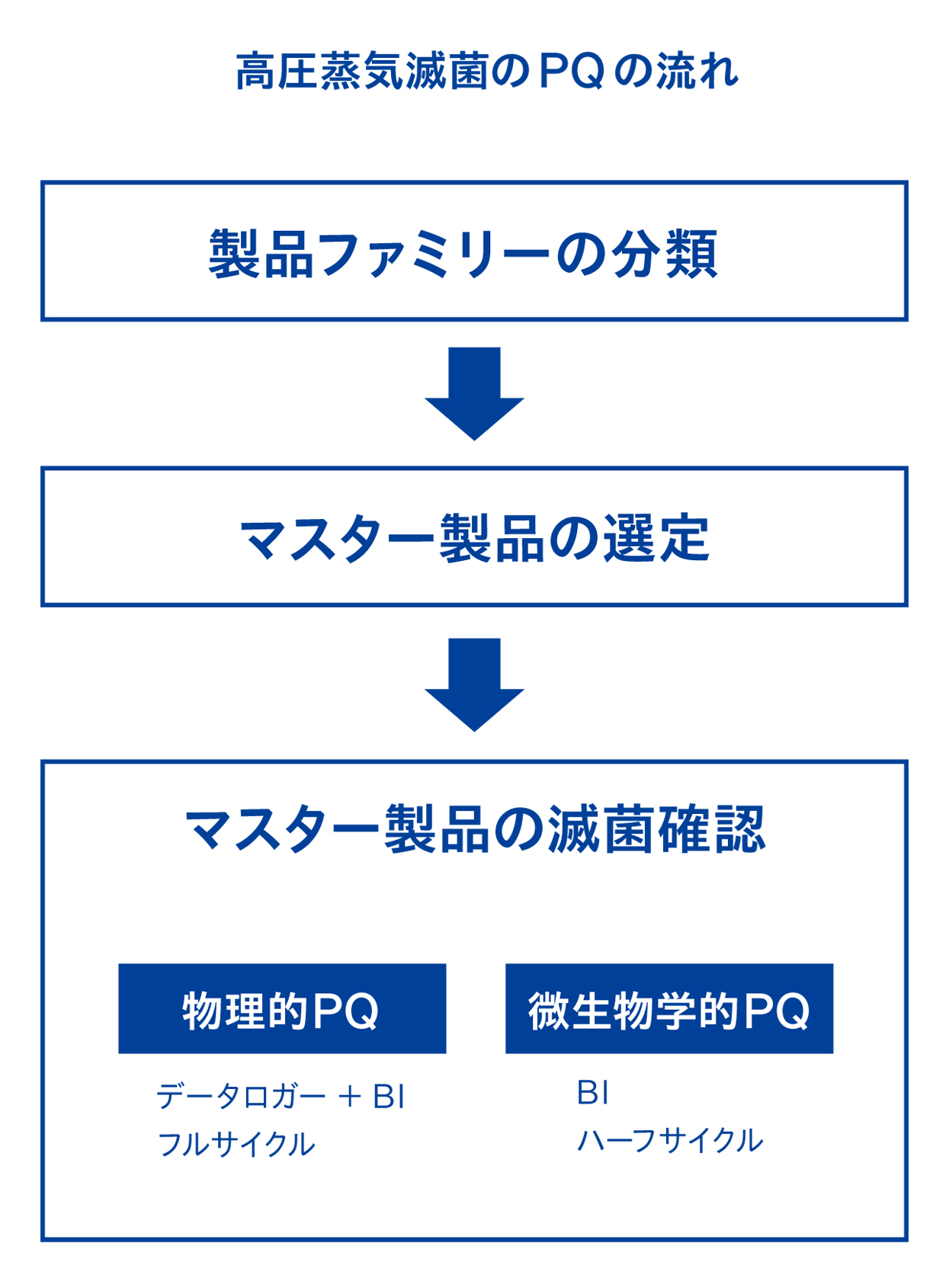
2-2. 製品ファミリーの分類:同じ条件で滅菌する製品群に分けること
製品ファミリーの分類は、各器材の添付文書や取り扱い説明書に記載されている滅菌条件を参考に、同じ条件で滅菌する製品群に分けることを指します。
例えば、ある器材の添付文書に「高圧蒸気滅菌の場合、134℃ 3分で滅菌する」と記載されていた場合、同じ滅菌条件(高圧蒸気滅菌、134℃ 3分)が記載されている器材と同じ製品ファミリーとします。
2-3. マスター製品の選定:製品ファミリーの中で最も滅菌しづらい製品を特定すること
製品ファミリー内のマスター製品を選定するには、包装毎に①空気排除の抵抗性、②器材の材質、③包装形態、④重量の4つの観点から点数をつけていきます。
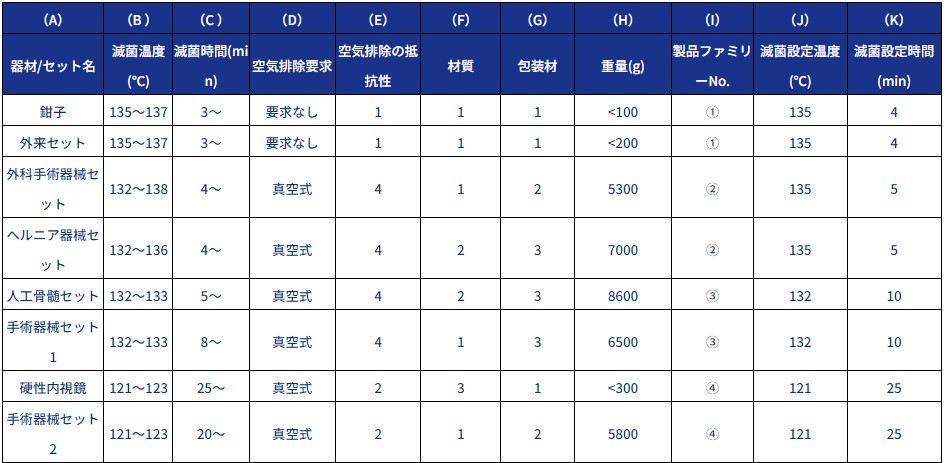
総合点が高いものをマスター製品としますが、同一製品ファミリー内で複数のマスター製品が存在した場合、それらもマスター製品候補として全て滅菌確認を行います。
製品ファミリーの分類、マスター製品の選定については、下記の記事もぜひご参考ください。
(記事)【第1種滅菌技師が解説】マスター製品と日常出荷判定用PCDの滅菌抵抗性を比較する方法
2-4. 実際にマスター製品を選定した例
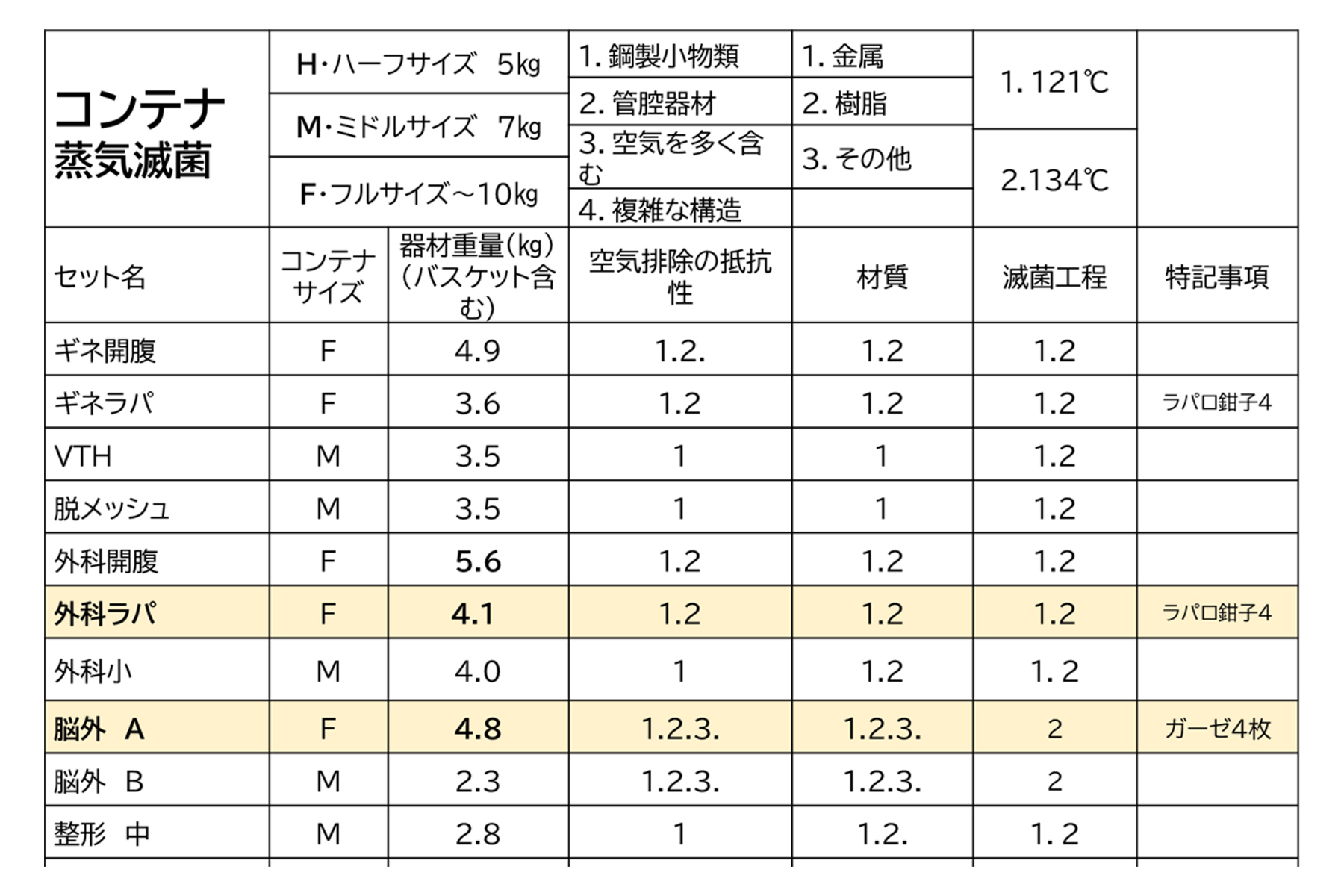
下記に実際の医療機関で行われたマスター製品の選定の例を紹介します。
製品ファミリーとして分類されたセットに点数をつけてみると下記の表になりました。
この場合、「外科ラパ」や「脳外 A」などがマスター製品候補になりますので、これらの滅菌確認を行います。
 マスター製品の候補になる「ラパロ鉗子セット」
マスター製品の候補になる「ラパロ鉗子セット」
2-5. マスター製品の滅菌確認:物理的PQもしくは微生物学的PQを行う
マスター製品候補が揃ったら、実際に滅菌ができているかどうかを確認するために、物理的PQ、または微生物学的PQのいずれかの方法で実施します。次章より物理的PQと微生物学的PQについて解説します。

3. 物理的PQとは
3-1. 実際の滅菌時間で処理した時の温度変化とBIの判定結果をもとに器材の滅菌保証を行う
物理的PQでは、実際の滅菌時間で処理したときのマスター製品の温度測定と、BIの死滅の確認をすることで器材の滅菌保証を行います。
この検証は、最大積載・最小積載の状態を3回ずつ実施する必要があります。一般的に、最大積載と最小積載の状態が滅菌しづらいとされているため、その両方でマスター製品の滅菌保証をしておくことで、その中間の積載量の検証を省くことができます。
3-2. データロガーを使用して器材内部の温度を測定する
物理的PQでは、データロガーと呼ばれる温度や圧力を測定できる機械を使用します。長い端子が付いたデータロガーを使用することで、写真のように器材内部の温度も測定することができます。

データロガーで黒い器材の内腔の温度を測定している様子
3-3. 器材内部の温度は、滅菌器の温度よりも遅れて上昇することがある
実際に器材の内部やコンテナ内部の温度を測定すると、滅菌器の温度と比較して遅れて温度上昇することがあります。この現象を「昇温遅れ」と呼びます。
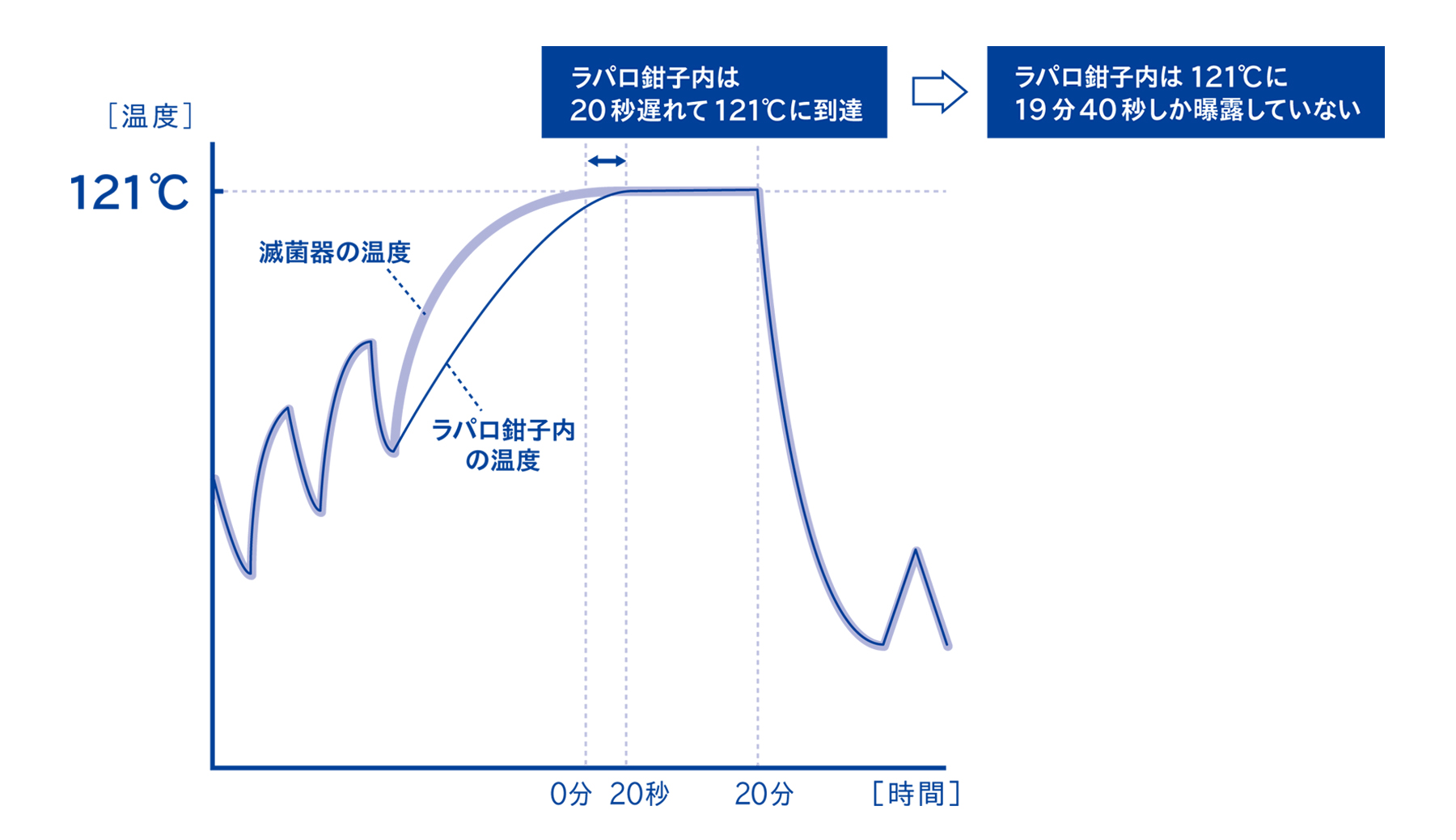
上の図は、器材内部の昇温遅れが20秒ほど観測されている例です。添付文書に記載されたこの器材の滅菌条件が121℃ 20分であった場合、器材内部は121℃ 19分40秒しか達成していないことになり、規定の条件に達していない可能性があります。
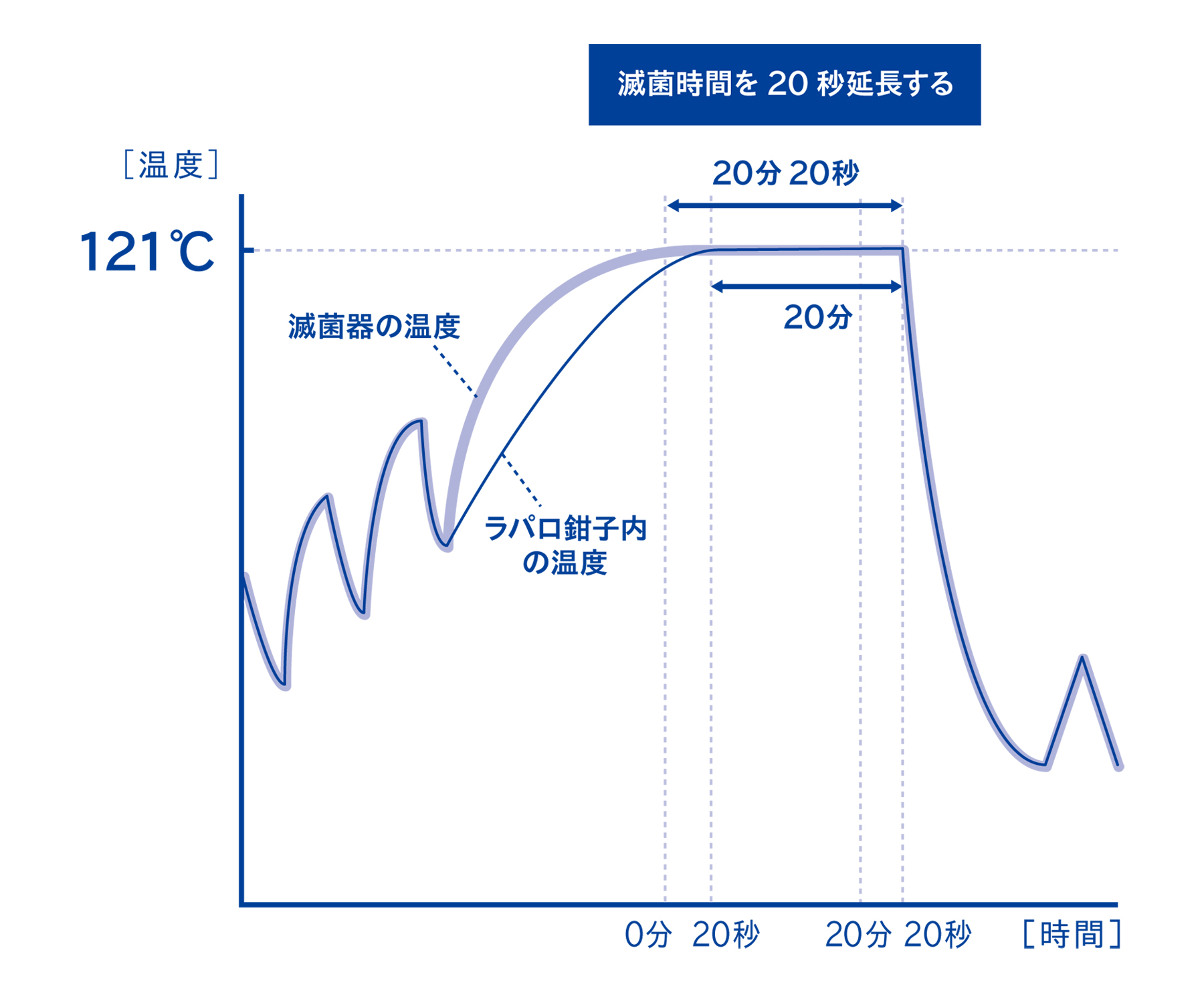
この昇温遅れによる滅菌不良の可能性を解消するためには、滅菌時間を延長する方法があります。この例であれば、滅菌時間を20分20秒にすることで、器材内部の滅菌時間を規定の20分にすることができるようになります。
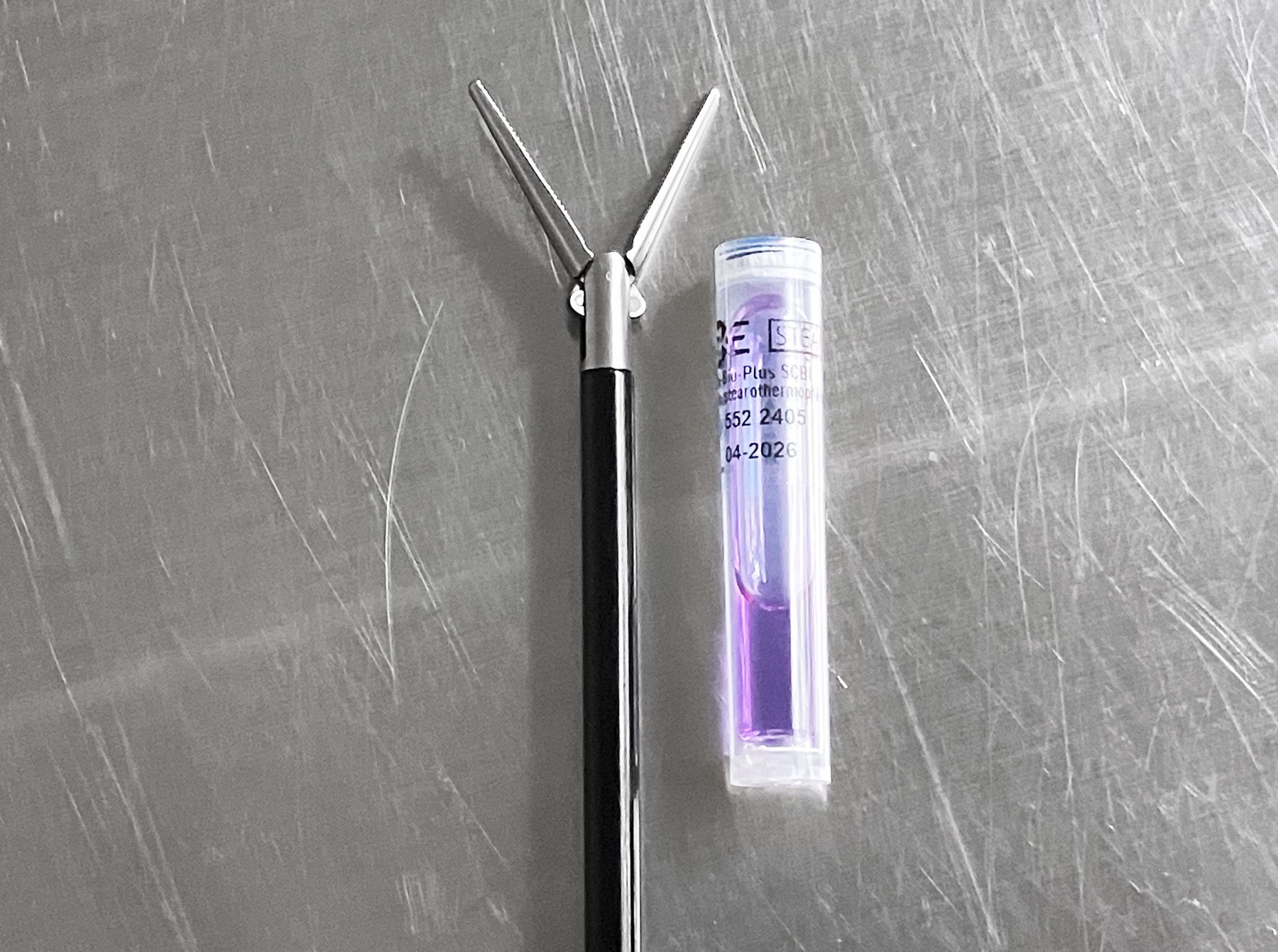
3-4. BIを包装材内部に設置する
データロガーを使用した温度測定の他に、BIを使用して菌の死滅を確認します。『ガイドライン2021』では、包装材内部にBIを設置すると記載されています。器材の横においた場合、器材内部の条件とは異なる点に注意しましょう。
3-5. データロガーを使用するPQは高額で、医療現場ではなかなか実施できないのが現状
以上のように、物理的PQは器材内部の温度情報を確認することのできる滅菌工程のバリデーションです。しかし、物理的PQに使用するデータロガーは高額であるため、医療現場では購入できないという声をよく伺います。
もしデータロガーを所持している滅菌器メーカーに借りることができたとしても、数日かかるPQの作業中はサービスマンの作業費がかかってくるため、こちらもまとまった予算が必要になります。
したがって医療現場ではなかなか実施できないというのが現状です。
4. 微生物学的PQ(通称: ハーフサイクル法)とは
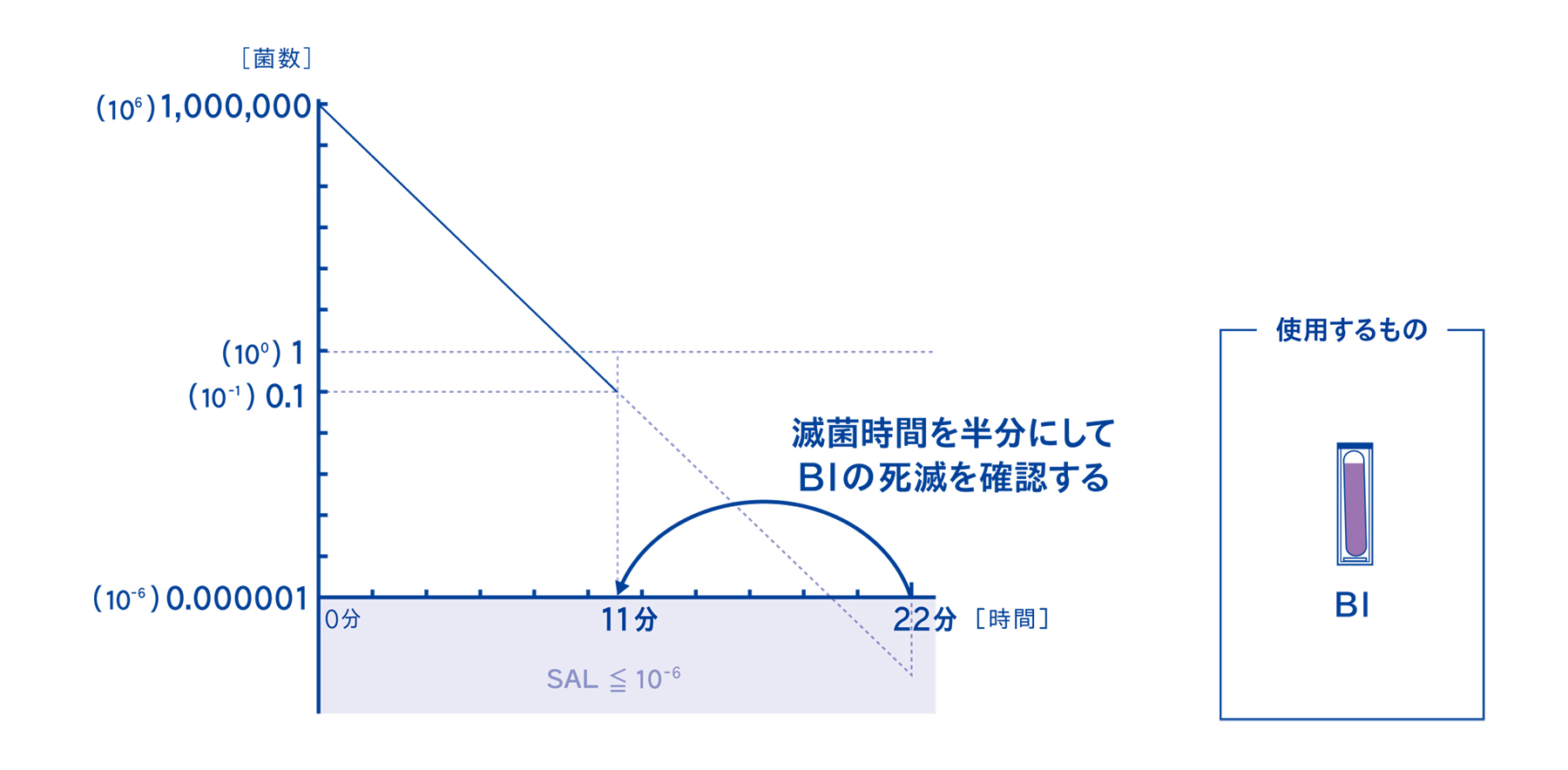
4-1. 実際の半分の滅菌時間で処理した時のBIの判定結果をもとに器材の滅菌保証を行う
微生物学的PQでは、実際の滅菌時間の半分の時間で処理した時のBIの判定結果を確認する方法です。高額なデータロガーを使用しないため、医療現場でも比較的実施しやすい方法といえます。
微生物学的PQでも物理的PQと同様に、最大積載・最小積載の状態で検証を3回ずつ実施する必要があります。
一般的に最大積載と最小積載の両方の状態が滅菌しづらいとされているため、その両方でマスター製品の滅菌保証をしておくことで、その中間の積載量の検証を省くことができます。
4-2. データロガーを使用せず、BIのみを使用して器材の滅菌保証をする
ハーフサイクル法ではデータロガーは使用せず、BIを使用して菌の死滅を確認します。データロガーを使用しない代わりに、実際の滅菌時間の半分の時間でBIの死滅を確認することで、実際の滅菌時間で滅菌した時に滅菌条件を達成するように工程を設計します。
微生物学的PQ(ハーフサイクル法)の例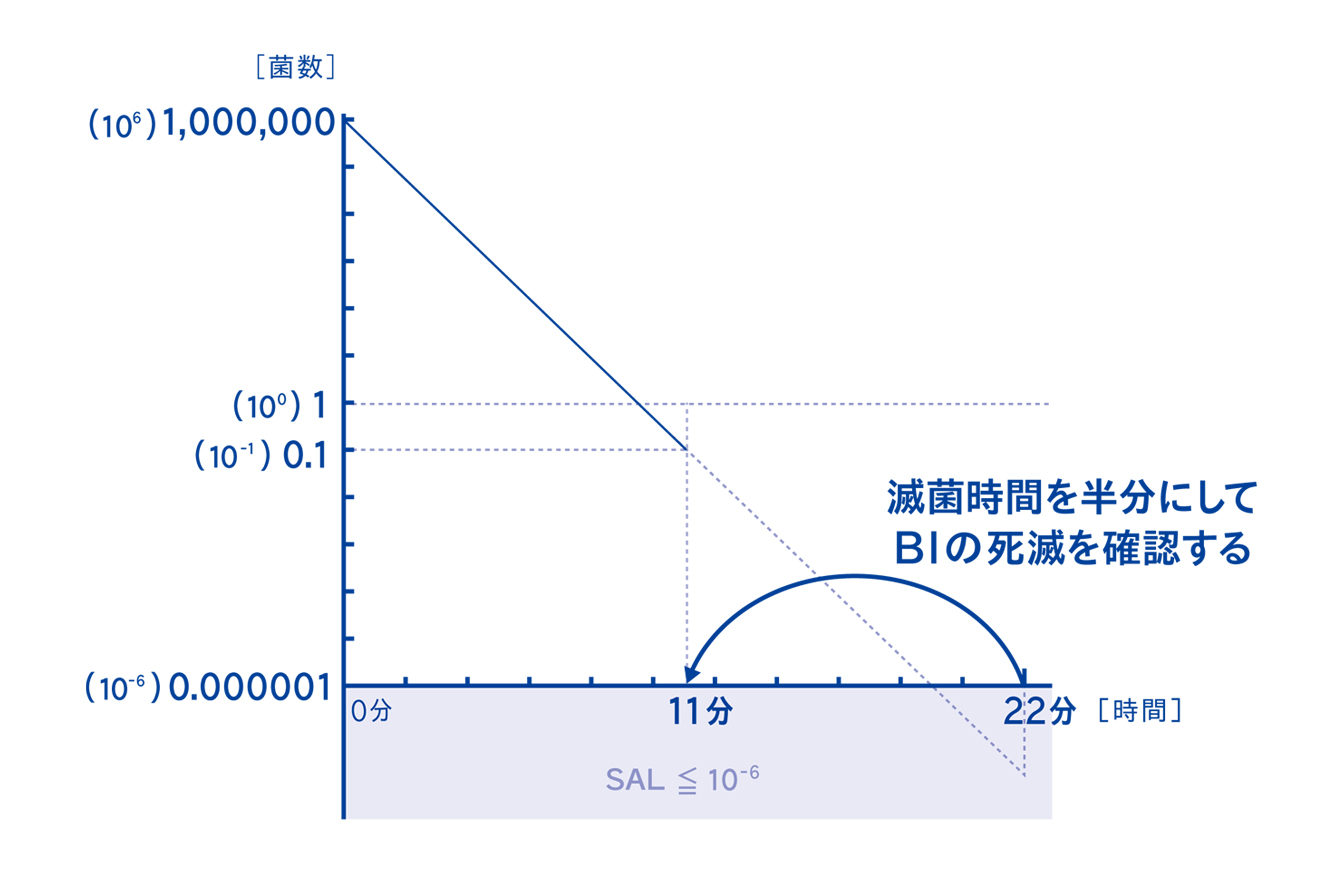
4-3. 微生物学的PQの課題は、BIを器材内部に設置できないこと
『医療現場における滅菌保証のガイドライン2021』では、「微生物学的PQにおいて、BIを包装内部に設置する」と記載されています。そして、物理的PQとは異なり、器材内部の滅菌条件の達成を確認しているわけではないことが注記されています。
BIは、設置された位置の情報しか得ることができません。器材の横に置いた場合、その器材の横が滅菌条件を達成したかがわかるだけで、器材内部の情報は何も得られません。
器材内部が滅菌条件を達成したかを知るには、器材内部にBIを設置する必要があります。しかし、細く繊細な内腔器材にはBIを設置することができません。このことが微生物学的PQの課題でした。
4-4. バイオコンパクトPCDで疑似的にBIを器材内部に設置する
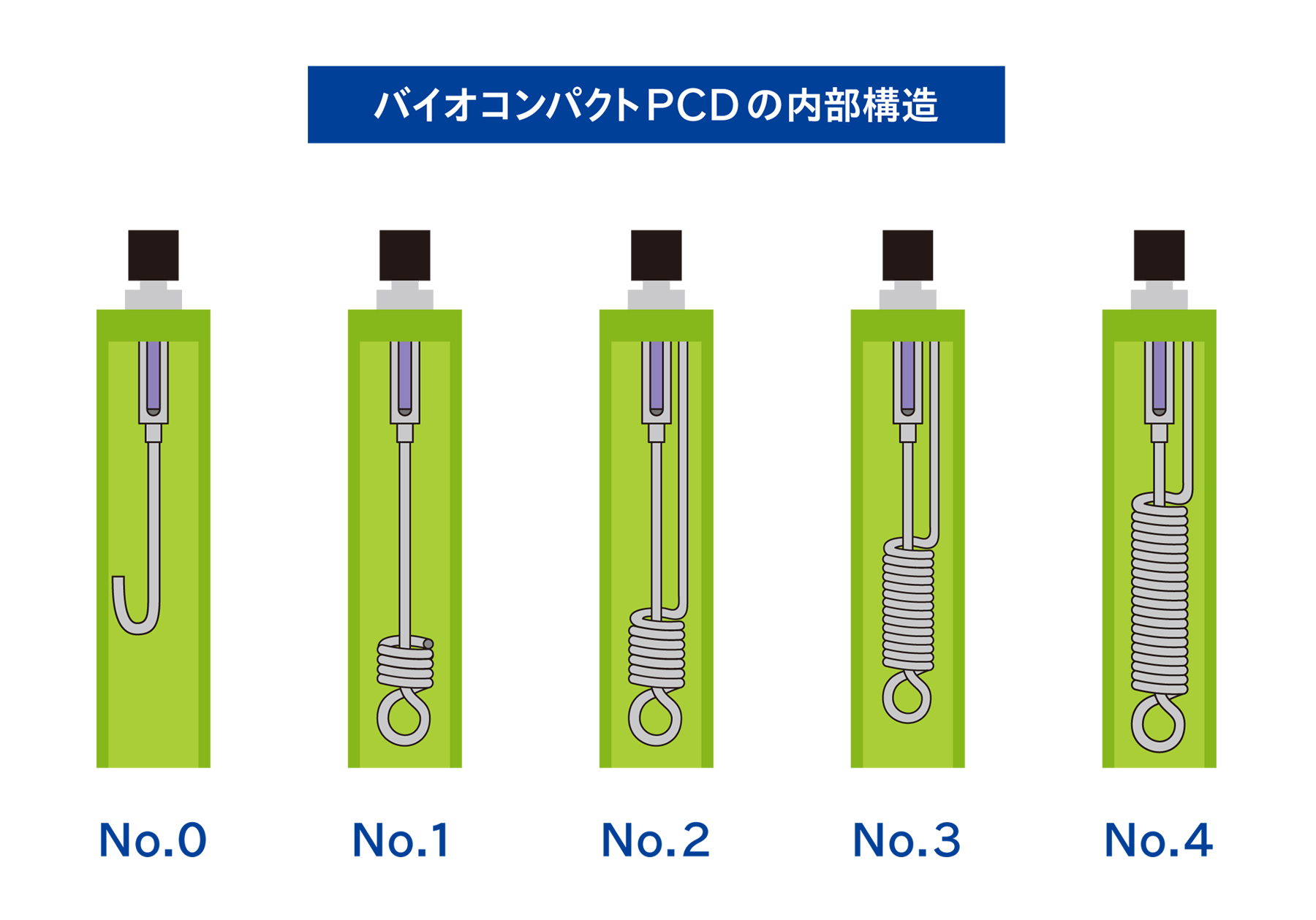
バイオコンパクトPCDは、BIを器材内部に設置することを疑似的に行えるPCDです。内部にBIを入れることができ、器材内部と同等以上の滅菌抵抗性を再現します。このバイオコンパクトPCDを使用することで、器材内部を含めた微生物学的PQを行うことができます。

バイオコンパクトPCDは、滅菌抵抗性の異なる5種類があります。マスター製品よりも滅菌抵抗性の高いPCDを選択することができるため、より精度の高い微生物学的PQを行うことができるようになります。
5. バイオコンパクトPCDを使用した微生物学的PQの事例
5-1. マスター製品は重量11.3kgのコンテナに入れた疑似ラパロ鉗子
ここで、バイオコンパクトPCDを使用した微生物学的PQの事例を紹介します。
今回の検証で使用するマスター製品はAAMI ST8 チャレンジPCDを参考にして作製しました。実際の器材セットの中には内腔器材が含まれていることが多いため、疑似ラパロ鉗子を加えて、更に負荷をかけたマスター製品とすることにしました。疑似ラパロ鉗子には内径8mm、長さ37.5cmのステンレス製の金属筒を使用しています。

なお、本検証では疑似的にマスター製品を作成していますが、基本的には実際の器材の中からマスター製品候補を選定し、その検証を行います。時間や予算の都合などで複数の検証を進めるのが困難な場合に、このように疑似的にマスター製品を作成することがあります。
5-2. 滅菌時間は規定の半分の時間で、最大積載・最小積載を3回ずつ実施する
疑似ラパロ鉗子の入った滅菌コンテナをマスター製品とし、滅菌温度は134℃、滅菌時間は半分の4分で処理しました。
なお、複数の滅菌条件がある場合は、それぞれの滅菌条件について最大積載・最小積載の場合を3回ずつ実施する必要があります。
5-3. コンテナ内の疑似ラパロ鉗子の横に、BIの入ったバイオコンパクトPCD(No.1)を置いて滅菌
下の写真のように、コンテナ内のラパロ鉗子の横に、BIの入ったバイオコンパクトPCDを置いて滅菌します。

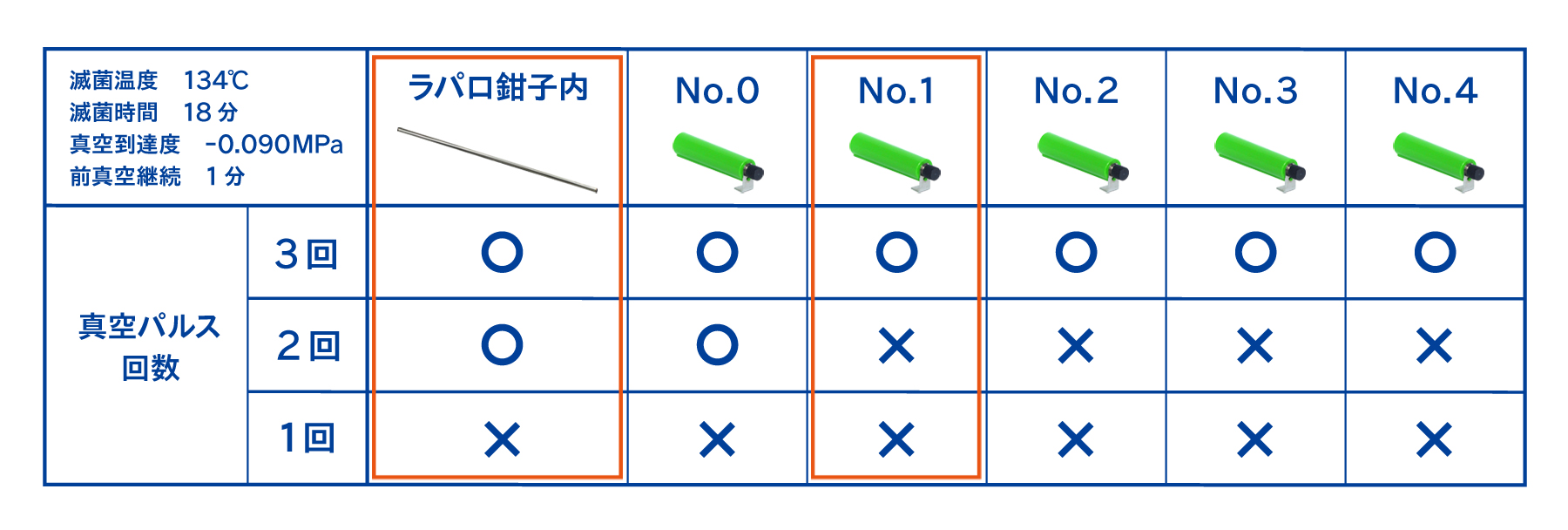
5-4. バイオコンパクトPCD(No.1)は疑似ラパロ鉗子より滅菌抵抗性が高い
事前検証により、滅菌抵抗性が疑似ラパロ鉗子<バイオコンパクトPCD(No.1)であることを確認しています。そのため今回の検証ではバイオコンパクトPCDのうち、No.1を選択しています。
なお、上記の器材とバイオコンパクトPCDの滅菌抵抗性の比較は、実際に滅菌する条件よりも悪条件にしたときに、器材内部とバイオコンパクトPCDの内部に設置したBIの結果を比べるという方法を用いています。
器材内部にはストリップ型BI、バイオコンパクトPCDの中には培地一体型BIを使用して比較することができるようにしています。ストリップ型BIを使用する場合、無菌操作が必要になります。バイオコンパクトPCDの選定については、SALWAYの再生処理アドバイザーにご相談ください。
ストリップ型BIの取り扱いの様子

5-5. バイオコンパクトPCDの中には10⁶菌のBIを設置する
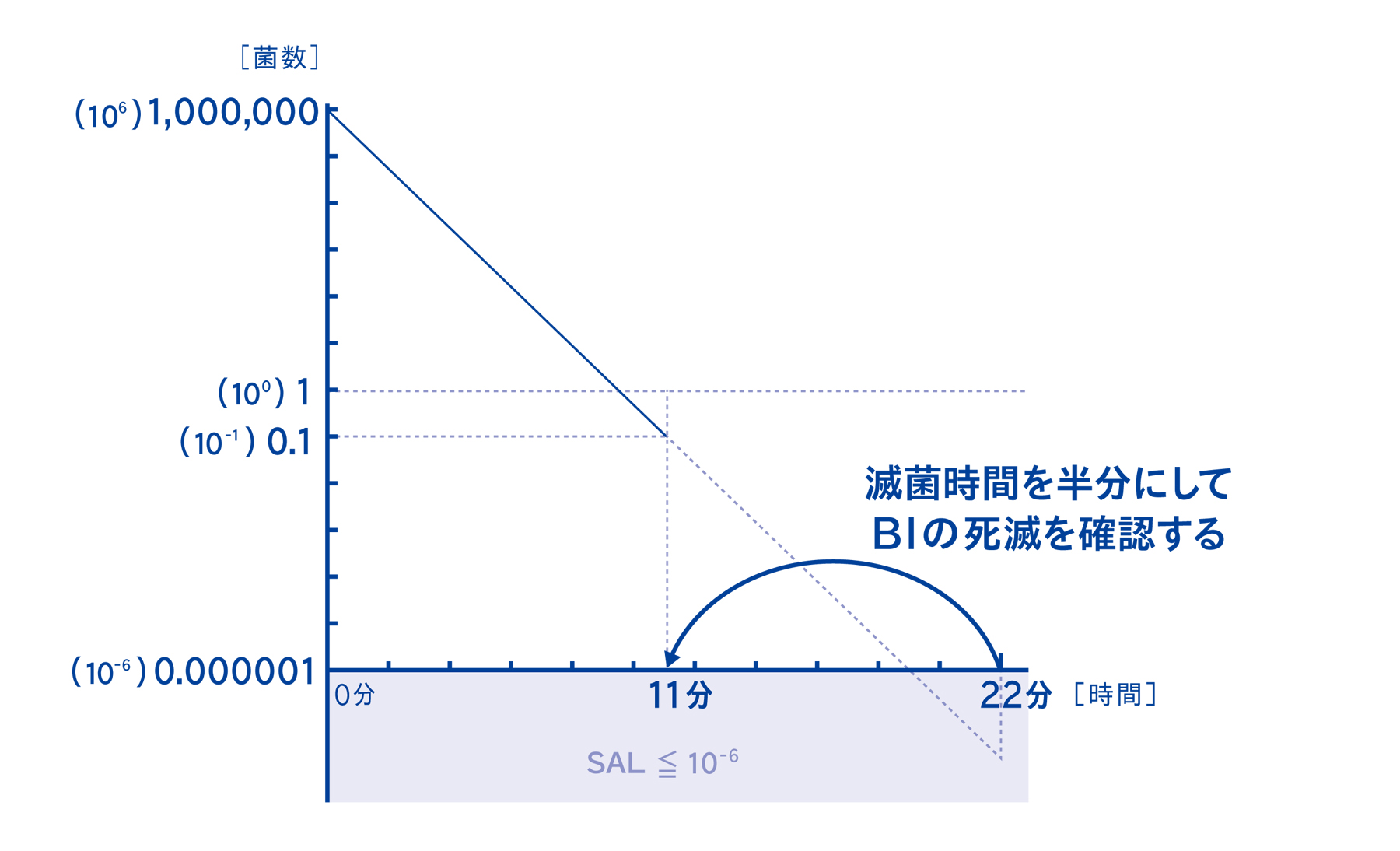
微生物学的PQの考え方は、初発菌数が10⁶菌、つまり100万個~1000万個の菌が存在するBIを基準として考えます。このBIが死滅する時間の倍の時間で滅菌することにより、SAL≦10-⁶を確実に達成するという考え方です。したがって、今回の検証では10⁶菌を使用しています。
なお、市販されているBIの中には10⁵菌、すなわち10万個~100万個の菌が存在するBIもあります。
5-5. 実際の培養結果
実際の半分の時間でコンテナ内に設置したバイオコンパクトPCDを滅菌した後、BIを培養した結果を下記に示します。いずれも合格していたため、実際の時間で滅菌した際に器材内部までSAL≦10⁻⁶が達成できる滅菌工程であることがわかりました。
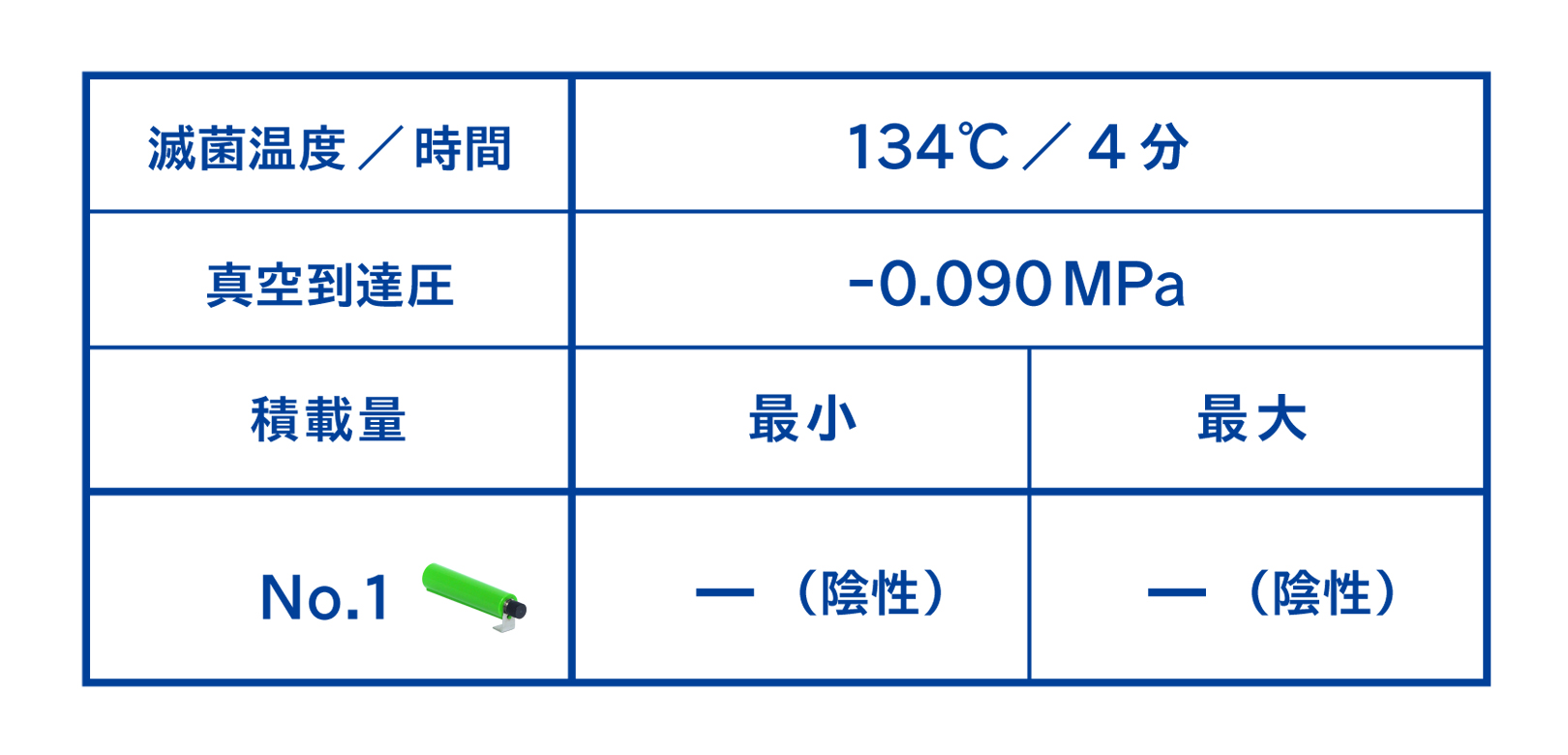
5-6. 不合格の時は真空工程の調整や、滅菌時間の延長などの対応をとる
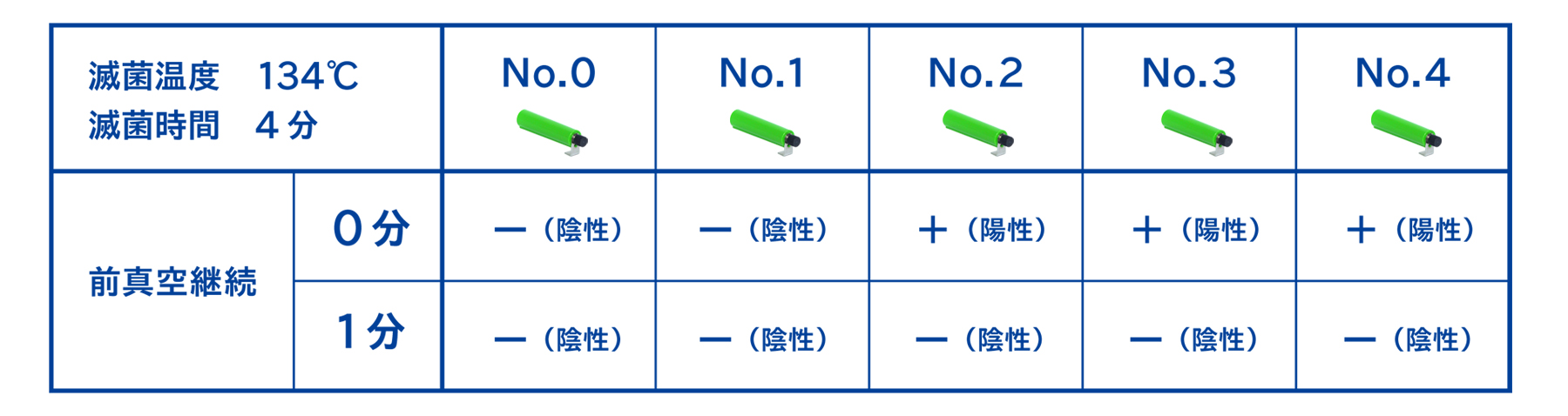
実際の半分の時間で滅菌した際にBIが不合格を示した場合は、真空工程の調整や滅菌時間の延長などの対策をとります。
例えば、下記の例では真空工程を改善させるために前真空継続時間*を延長したところ、合格するバイオコンパクトPCDがNo.1からNo.4までに改善しました。
前真空継続時間とは、設定した真空到達度に達してから真空ポンプを継続して回転させる時間のことです。設定した真空到達度よりも高い空気除去が望めます。
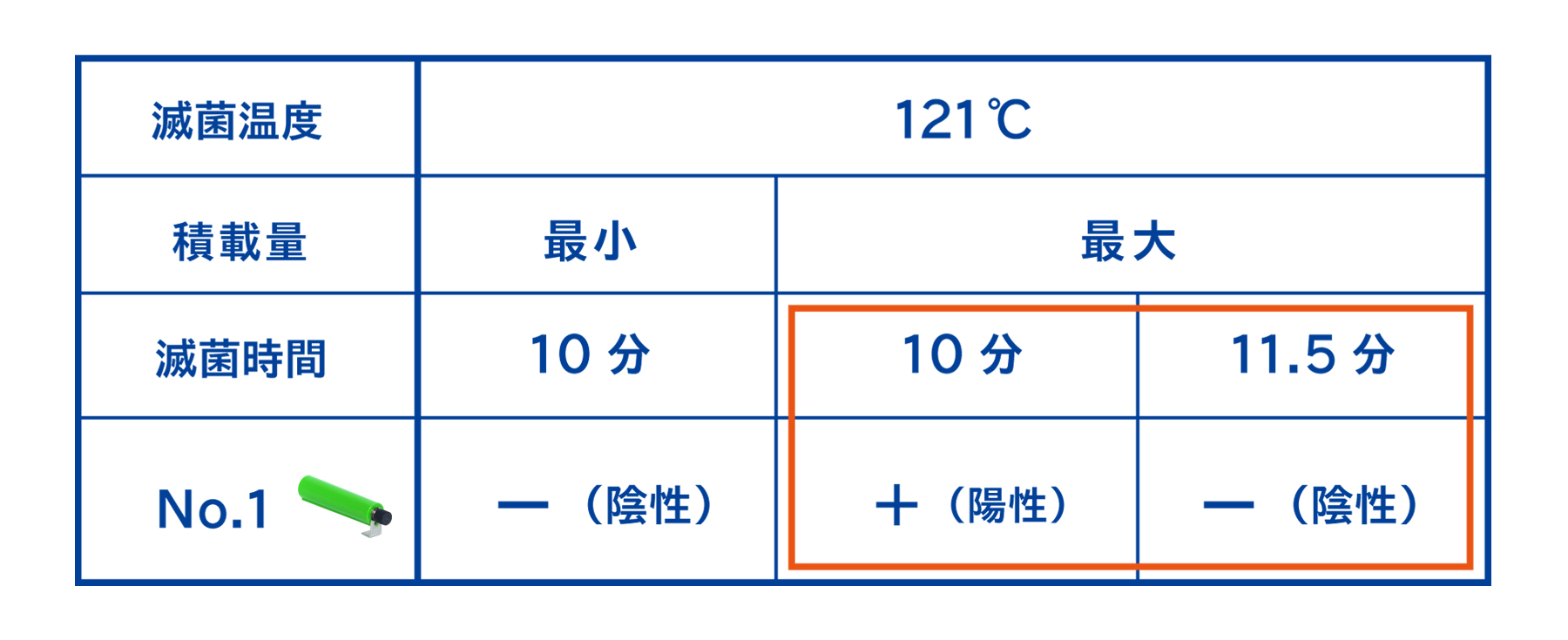
また、下記のように滅菌時間の延長をすることで改善させることができる場合があります。こちらのケースでは、121℃ 10分で滅菌したところBIは陽性を示したため、121℃ 11.5分に延長したところBIが陰性を示しました。したがって実際の滅菌時間を23分に設定することで改善させることができました。
6. まとめ
いかがでしたでしょうか。
安全な器材の払い出しをするためには、再生処理の全ての工程のバリデーションが必要です。滅菌工程のバリデーションのうち、IQ・OQは滅菌器メーカーが、PQ(マスター製品の滅菌確認)は中央材料室が実施する必要があります。PQの方法は2種類あり、物理的PQと微生物学的PQがあります。物理的PQは費用が高額であること、微生物学的PQは器材内部にBIを設置できないことが課題でした。
本記事で紹介したバイオコンパクトPCDを用いた微生物学的PQは、器材内部にBIを設置できないという課題を解決するものです。バイオコンパクトPCDは器材内部と同等以上の滅菌抵抗性を持っており、器材内部でBIが死滅するかどうか検証することができます。
本記事の内容に関するお問合せは、営業担当またはSALWAYウェブサイトのお問合せフォームよりご連絡下さい。